Keratosis Follicularis Spinulosa Decalvans : A New Observation
Elmahi H1*, Elloudi S1, Gallouj S1, Mernissi FZ1, Rimani M2
1 Department of Dermatology, University Center, Fez, Morocco.
2 Anatomy - Pathological Center Hassan, Rabat, Morocco.
*Corresponding Author
Hakima Elmahi,
Department of Dermatology,
University Center, Fez, Morocco.
E-mail: elmahi.hakima@gmail.com
Received: January 09, 2017; Accepted : February 09, 2017; Published: February 10, 2017
Citation: Elmahi H, Elloudi S, Gallouj S, Mernissi FZ, Rimani M (2017) Keratosis Follicularis Spinulosa Decalvans : A New Observation. Int J Clin Dermatol Res. 5(1), 107-109. doi: http://dx.doi.org/10.19070/2332-2977-1700027
Copyright: Elmahi H© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Keratosis follicularis spinulosa decalvans (KFSD) is a rare condition characterized by diffuse keratosis pilaris with a scarring alopecia of the scalp and associated photophobia, facial erythema, and palmoplantar keratoderma. Although initially described as a sex-linked disorder, several different inheritance patterns have been observed. We describe a patient whose mother and sister were also affected with this condition, consistent with an autosomal dominant genetic transmission. Multiple topical and systemic treatments have been unsuccessful in this patient, attesting to the treatment refractoriness typically seen in KFSD.
2.Introduction
3.Patient and Observation
4.Discussion
5.Conclusion
6.References
Keywords
Keratosis Pilaris Atrophicans; Keratosis Follicularis Spinulosa Décalvans; Autosomal Dominant.
Introduction
Scarring alopecia in association with follicular hyperkeratosis is the primary characteristic of keratosis pilaris atrophicans (KPA). It affects mainly the face and scalp and can be inflammatory in nature [1]. Widespread KP can also be seen. keratosis pilaris atrophicans is a group of cutaneous disorders that may represent a spectrum of 1 disease. Differences in localization and the degree of inflammation and atrophy have been used to distinguish these various disorders [1]. They are genetic disorders with different modes of inheritance. Heterogeneity in the mode of inheritance exists not only between different types of KPA but also within 1 type, such as keratosis follicularis spinulosa decalvans (KFSD) [1, 2]. The pathogenesis is not known, but abnormal follicular keratinization has been suggested.
Patient and Observation
A 45 -year-old male who complained of progressive hair loss over the scalp since 15 years and over the eyebrows since 7 years. He gave a history of similar but milder complaints being present in his mother and younger sister suggesting X-linked inheritance. On examination of the scalp, she had multiple patches of cicatricial alopecia with sparse hair in the frontal, parietal, and temporal areas of the scalp [Figure 1 et 2]. Sparse hair over the eyebrows [Figure 2], eyelashes [Figure 2], axillae, and pubic area were noted. He also had generalized dry skin and multiple follicular papules over the trunk, upper; abdomen and lower limbs [Figure 3] and buttocks. Dermoscopic examination revealed cicatricial alopecia on the scalp, showing keratosis and adherent squamae [Figure 4]. There was no history of photophobia and ophthalmic examination showed no evidence of conjunctival/corneal involvement. Histopathologic examination revealed hyperkeratosis and mild acanthosis of epidermis with follicular plugging and dilated infundibulum [Figure 5]. The dermis revealed perifollicular lymphocytic infiltrate with eccentric epithelial atrophy, polytrichia, and perifollicular fibrosis [Figure 5 et 6]. With clinic pathological correlation, and Considering family history, a diagnosis of KFSD was offered. Multiple topical and systemic treatments have been unsuccessful in this patient, attesting to the treatment refractoriness typically seen in KFSD.
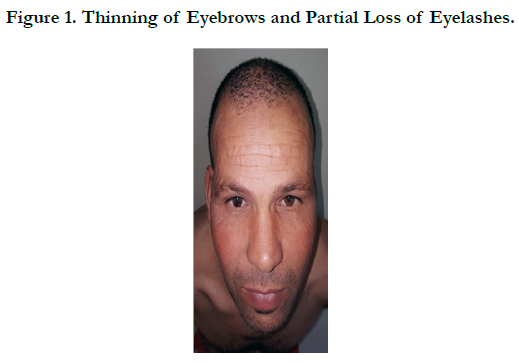

Figure 1. Thinning of Eyebrows and Partial Loss of Eyelashes.
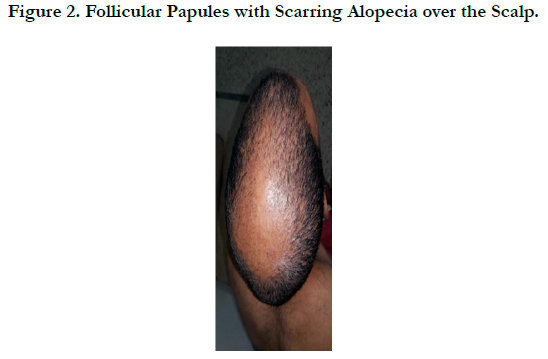
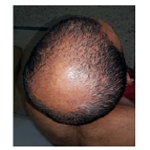
Figure 2. Follicular Papules with Scarring Alopecia over the Scalp.
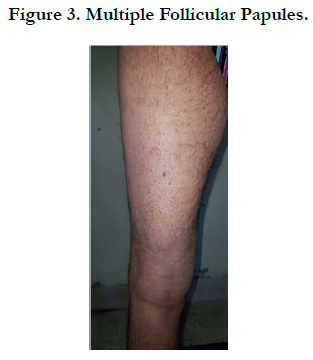
Figure 3. Multiple Follicular Papules.
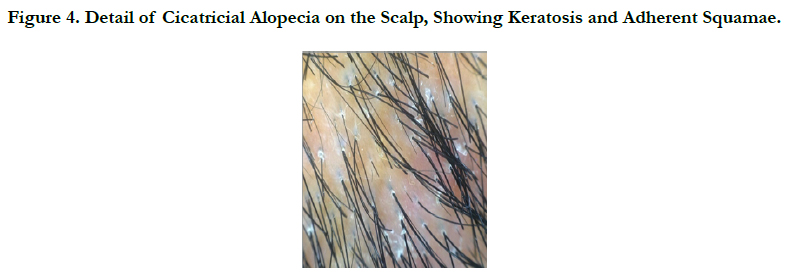
Figure 4. Detail of Cicatricial Alopecia on the Scalp, Showing Keratosis and Adherent Squamae.
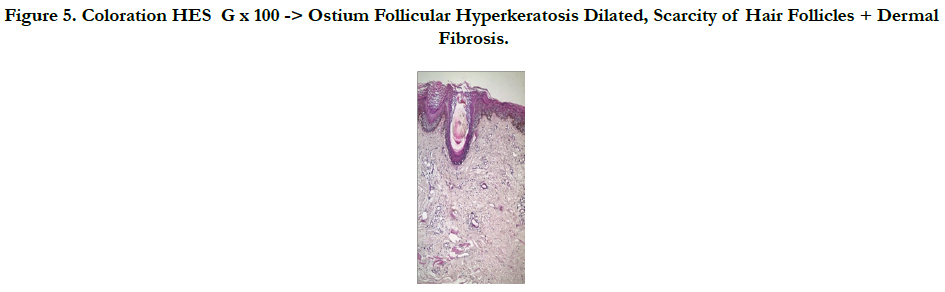
Figure 5. Coloration HES G x 100 -> Ostium Follicular Hyperkeratosis Dilated, Scarcity of Hair Follicles + Dermal Fibrosis.
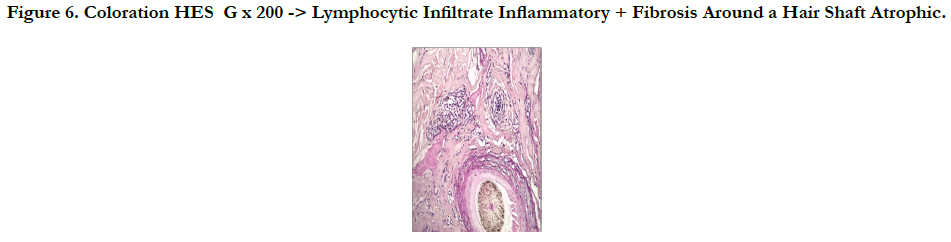
Figure 6. Coloration HES G x 200 -> Lymphocytic Infiltrate Inflammatory + Fibrosis Around a Hair Shaft Atrophic.
Discussion
keratosis follicularis spinulosa decalvans is one of several related disorders that are distinguished by the presence of keratosis pilaris with inflammation and subsequent atrophy. These disorders have been grouped under the category of keratosis pilaris atrophicans, which comprises [1-3]. KFSD (involvement of scalp, eyebrows, and eyelashes in addition to other associated features such as palmoplantar keratoderma, photophobia, widespread KP, and atopic); Keratosis pilaris atrophicans facial (KPAF) or ulerythema ophryogenes (involvement of eyebrows, forehead, and cheeks) ; Atrophoderma vermiculatum (AV) (involvement of the cheeks and periauricular regions, which is followed by reticulate atrophy) the variant of KFSD that starts at puberty is called folliculitis spinulosa decalvans (FSD).
Siemens first used the term KFSD in 1926 to describe findings in a Bavarian family whose members were affected by diffuse generalized follicular papules, alopecia, and loosening of the scalp, eyebrow, and eyelash hairs [2]. Associated findings in this kindred included photophobia, corneal and conjunctival inflammation accompanied by tearing, and hyperkeratotic lesions of the palms and soles. While the onset of ophthalmitis, photophobia, and tearing is described typically within the first few weeks or months after birth, keratosis pilaris and scalp alopecia are typically delayed until puberty or thereafter. An X-linked inheritance pattern has been observed, with the mutation present at Xp22.1, encoding the spermidine/spermine N(1)-acetyltransferase (SSAT) gene, and in keeping with this, males are generally noted to be more severely affected than females. However, the condition has also been seen in families in which the pattern of transmission suggests an autosomal dominant pattern, as in the family presented herein [2]. KFSD results from mutations in the MBTPS2 gene. Mutations in this gene also underlie ichthyosis follicularis, alopecia and photophobia syndrome [4]. The differential diagnosis of KFSD include icthyosis follicularis alopecia photophobia (IFAP) syndrome (which is characterized by non-scarring alopecia), lichen planopilaris, and lichen spinulosus. IFAP syndrome is characterized by non-scarring alopecia in contrast to KFSD. The other two conditions can be also be differentiated by thorough clinic-pathological examination [5]. There is no specific treatment for KFSD. Various drugs have been tried to delay scarring alopecia such as isotretinoin [6] and dapsone [7]. For symptomatic relief emollients, topical steroid, and keratolytic agents can be used [4]. Different systemic treatments including isotretinoin, etretinate, dapsone and antibiotics have been tried with varied results it is suggested that the use of retinoids in the early and more active phase of the disease, when histopathology shows perifollicular infiltrate, can bring some benefit [4, 6, 7]. Treatment is even more disappointing when the disease predominantly shows cicatricial changes, as in this present study [8]. Treatment in this circumstance is reducedonly to the use of topical palliative medication [8].
Conclusion
Although it is a rare genodermatosis, KFSD should always be considered in all cases of hyperkeratosis with alopecia as that apart from the genetic counseling needed in some cases, the treatment of such disease should be started the earliest possible (ideally still in the inflammatory phase) so as to retard and minimize the cicatricial sequels [8].
References
- Alfadley A, Al Hawsawi K, Hainau B, Al Aboud K (2002) Two brothers with keratosis follicularis spinulosa decalvans. J Am Acad Dermatol. 47(5):S275-8.
- Bellet JS, Kaplan AL, Selim MA, Olsen EA (2008) Keratosis follicularis spinulosa decalvans in a family. J Am Acad Dermatol. 58(3): 499-502.
- Rand R, Baden HP (1983) Keratosis Follicularis Spinulosa Decalvans: Report of Two Cases and Literature Review . Arch Dermatol. 119(1): 22-26.
- Zhang J, Wang Y, Cheng R, Ni C, Liang J, et al., (2016) Novel MBTPS2 missense mutation causes a keratosis follicularis spinulosa decalvans phenotype: mutation update and review of the literature. Clin Exp Dermatol. 41(7): 757-60.
- Malvankar DD, Sacchidanand S (2015) Keratosis Follicularis Spinulosa Decalvans: A Report of Three Cases. Int J Trichology. 7(3): 125-8.
- Gupta D, Kumari R, Bahunutula RK, Thappa DM, Toi PCh, et al., (2015) Keratosis follicularis spinulosa decalvans showing excellent response to isotretinoin. Indian J Dermatol Venereol Leprol. 81(6): 646-8.
- Kunte C, Loeser C, Wolff H (1998) Folliculitis spinulosa decalvans: Successful therapy with dapsone. J Am Acad Dermatol. 39(5 Pt 2): 891-3.
- Berbert AL, Mantese SA, Rocha A, Cherin CP, Couto CM (2010) Keratosis follicularis spinulosa decalvans - Case report. A Bras Dermatol. 85(4): 537-40.