Strategies for the use of SPE
Hideharu Shintani1
1 Chuo University, School of Science, Kasuga Bunkyo, Tokyo, Japan.
*Corresponding Author
Hideharu Shintani
Chuo University,
School of Science,
Kasuga Bunkyo, Tokyo, Japan.
Tel: +81425922336
E-mail: shintani@mail.hinocatv.ne.jp
Article Type: Research Article
Received: August 12, 2013; Accepted: August 29, 2013; Published: August 30, 2013
Citation: Hideharu Shintani (2013) Strategies for the use of SPE. Int J Clin Pharmacol Toxicol. 2(6), 87-96. doi: dx.doi.org/10.19070/2167-910X-1300017
Copyright: Hideharu Shintani© 2013. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
2.Introduction
3.Solid Phase Extraction (SPE) as a Filter
3.1.Materials and Methods for GHB Analysis
3.1.1.Reagents
3.1.2.Standards and Solutions
3.1.3.Sorbent
3.1.4.Instrumentation
3.2.GC-MS Analysis
3.3.Urine GHB Extraction Procedure Using a ZS
3.4.Results
3.4.1.Chromatography
3.4.2.Recovery
3.4.3.Linearity
3.4.4.Stability
3.4.5.Sensitivity
3.4.6.Interferences
3.5.Summary
4.SPE-Selective Adsorption
4.1.Gabapentin in Serum, Plasma, or Whole Blood for GC or GC-MS Analysis Using a 200-mg CLEANUP C18 Extraction Column
4.2.Assay Validation
4.3.Summary
5.SPE in Copolymeric Interaction
5.1.General GC-MS Method
5.2.Forensic Drug Analysis for GC or GC-MS Using a 200-mg CLEAN SCREEN Extraction Column (ZSDAU020 or ZCDAU020)
5.3.The Future of SPE
6.References
Keywords
SPE; GHB; Pharmaceutical Compounds; GC-MS.
Introduction
In this paper we discuss four basic strategies used in solid phase extraction
(1) filtration,
(2) selective adsorption,
(3) copolymeric extraction, and
(4) immunoaffinity.
Most extractions will fall into one of these categories.
Solid Phase Extraction (SPE) as a Filter
One of the first uses of SPE was as a filtration device in pharmaceutical laboratories where scientists were extracting active compounds from fermentation broths. SPE cartridges were used to remove as much of the matrix as possible, allowing active compounds to pass through the columns and to be collected. This technique has evolved into a more sophisticated largescale type of chromatography, called flash chromatography, used by organic synthetic chemists. Flash chromatography has not found a place in forensic toxicology as yet. Figure 1 shows a graphic demonstration of this type of approach to SPE. The example shown is the isolation of γ-hydroxybutyric acid (GHB) from human urine. Urine samples are filtered by a special sorbent that holds interfering substances and allows the analytes of interest to pass through the column to be analyzed. In this case the biggest contaminant is urea, which is effectively removed by the SPE column. The following describes a forensic application for GHB from human urine that uses SPE as a filtering step followed by additional liquid-liquid extraction techniques.
GHB has become widely known as the “sex drug for the 1990s. In the new “mi1lenium, GHB has become popular with college students and club-goers as a mood modulator. Cases of GHB use were rare, but have become numerous over the last two years. On ingestion, it reduces inhibitions and reportedly increases libido. Other popular street names include “scoop” and “liquid ecstasy.” This drug has been classified as a date rape drug, along with lorazepam, ketamine, and flunitrazepam.
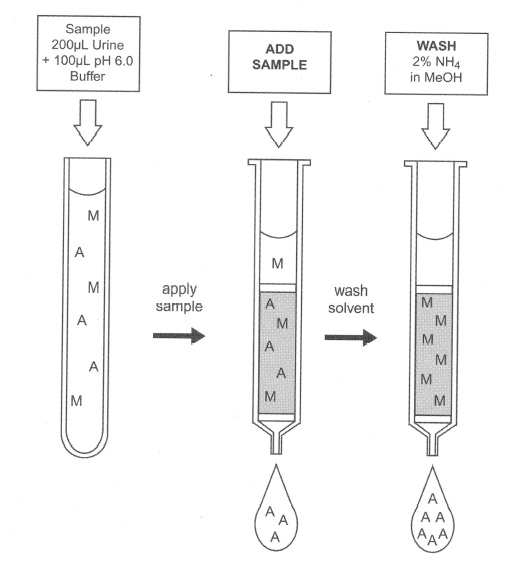
Figure 1: The use of an SPE column as a filter.
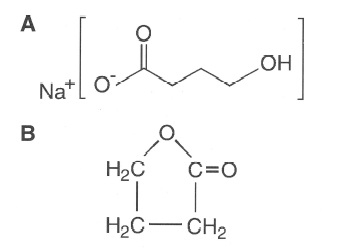
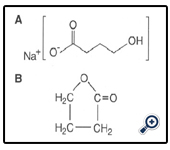
Figure 2: Chemical structures of (A) γ-hydroxybutyric acid and (8) butyrolactone.
GHB is an endogenous human metabolite structurHideharu Shintani, International Journal of Clinical Pharmacology & Toxicology 2013, 2:602 2 ally similar to the neurotransmitter γ-amino butyric acid (GABA) [1, 2]. It was first synthesized and used in Europe as an anesthetic, but was later discontinued because this drug was widely sold in health food stores as a weight control drug (functionally similar to L-tryptophan) and to induce the secretion of growth hormone for body building. In 1990 the sale and distribution of GHB was federally banned owing to its potentially harmful effects [3].
GHB is easily synthesized from γ-butyrolactone, a compound that is found in some commercial products. Figure 2 shows the chemical structures of GHB and butyrolactone. The Internet has popularized this drug by giving recipes on how to e manufacture it. One particular book claims the drug to be a “natural mood enhancer” [4]. In clandestine preparations the concentrations vary significantly, leading to problems with overdosage. Harmful contaminants from low-quality ingredients may also appear. In the 1990s more than 20 deaths in the United States were attributed to the of sort use of GHB, generally in conjunction with alcohol ingestion [5].
GHB is a small polar molecule that is very difficult to separate for a qualitative determination. In many methods it is extracted following conversion to butyrolactone and chemically derivatized by silylation [3, 6,7]. This method allows for the chemical derivatization of the parent compound without the formation of the Figure 1: The use of an SPE column as a filter Figure 2: Chemical structures of (A) γ-hydroxybutyric acid and (8) butyrolactone Hideharu Shintani, International Journal of Clinical Pharmacology & Toxicology 2013, 2:602 3 lactone. Instrumental analysis is performed using gas chromatography-mass spectrometry (GC-MS).
in Europe as an anesthetic, but was later discontinued because this drug was widely sold in health food stores as a weight control drug (functionally similar to L-tryptophan) and to induce the secretion of growth hormone for body building. In 1990 the sale and distribution of GHB was federally banned owing to its potentially harmful effects [3].
GHB is easily synthesized from γ-butyrolactone, a compound that is found in some commercial products. Figure 2 shows the chemical structures of GHB and butyrolactone. The Internet has popularized this drug by giving recipes on how to e manufacture it. One particular book claims the drug to be a “natural mood enhancer” [4]. In clandestine preparations the concentrations vary significantly, leading to problems with overdosage. Harmful contaminants from low-quality ingredients may also appear. In the 1990s more than 20 deaths in the United States were attributed to the of sort use of GHB, generally in conjunction with alcohol ingestion [5].
GHB is a small polar molecule that is very difficult to separate for a qualitative determination. In many methods it is extracted following conversion to butyrolactone and chemically derivatized by silylation [3, 6,7]. This method allows for the chemical derivatization of the parent compound without the formation of the lactone. Instrumental analysis is performed using gas chromatography-mass spectrometry (GC-MS).
Certified American Chemical Society (ACS)-grade hexane, sodium phosphate monobasic, sodium phosphate dibasic, dimethylformamide, ammonium hydroxide, and HPLC-grade methanol and ethyl acetate were all purchased from Mallinckrodt (Phillipsburg, PA). Distilled water was prepared using a millipore purification system, bis (trimethylsilyl) trifluoroacetamide (BSTFA) with 1 % trimethylchlorosilane (TMCS) from United Chemical Technologies, Inc. (Bristol, PA).
a. GHB sodium salt, was purchased from Aldrich (Milwaukee, WI).
b. GHB-D6 was purchased from Cerrilliant (Austin,TX). The GHB and GHB-D6 were prepared to 0.1mg/mL in methanol.
c. Prepare the 0.1 M phosphate buffer, pH 6.0, by dissolving 1.70 g of Na2HPO4 and 12.14g of NaH2PO4·H2O in 800 mL of DI H2O. Dilute to 1000 mL using DI H2O. Mix. The solution is stable for at least 1 month.
d. Adjust pH to 6.0 ±0.1 with 0.1 M monobasic or dibasic sodium phosphate.
e. The CH3OH-NH4OH (99:1) is prepared fresh daily.
The extraction columns were CLEAN SCREEN ZSGHB020 containing 200 mg of sorbent in a 10-mL column and were manufactured by United Chemical Technologies, Inc.
A Hewlett Packard 5971A Mass Selective Detector, a 7673 Autosampler, and a 5890 Gas Chromatograph fitted with a 30-m, 0.25 mm i. d. , 0.25μm film thickness Rtx-5 (comparable to a DB-5 or HP-5) were from Restek (Bellefonte, PA).
Injection Volume 1μL, Splitless injection Run time is extended past the elution of the GHB to eliminate any residual BSTFA or urine byproducts.
The method uses selected ion monitoring (SIM) for three ions for each analyte. The dwell times were set to 30 ms per ion, resulting in 3.62 cycles per second. The most prevalent ions for GHB-diTMS are 147, 233, 148, 149, 204, 143, and 234 m/z. The most prevalent ions for GHB-D6-diTMS are 147, 239, 148, 149, 206, and 240 m/z. Urea is also derivatized by BSTFA to form a diTMS derivative. It elutes near GHB and has many of the same ions including 147, 148, and 149; therefore, some of the less abundant ions must be used for the SIM analysis. Table 1 lists these ions.
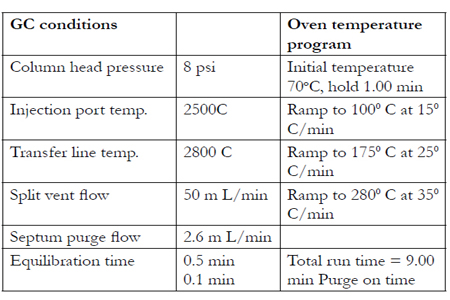

Table 1: Ions Using SIMS Analysis and Quantitation lon.
1. Sample preparation
a. a.To 200 μL of urine add internal standard (GHB·D6) and 100 μL of 0.1 M phosphate buffer, pH 6.0.
b. Mix/vortex.
2. Condition CLEAN SCREEN@GHB extraction column.
a. 1 x 3 mL of CH3OH; aspirate.
b. 1 x 3 mL of DI H2O; aspirate.
c. 1 x 0.5 mL 0.1 M phosphate buffer, pH 6.0;aspirate.
Note: Aspirate at ≦3 in. Hg to prevent sorbent drying.
3. Apply sample.
a. Place test tubes into vacuum manifold for collection.
b. Collect both the sample loading and wash.
c. Decant sample onto column. Aspirate at ~1 in. Hg.
4. Wash column
a. Add 1 mL of CH3OH-NH4OH (99:1) to sample test tube; vortex.
b. Decant wash onto column.
Note: Aspirate at ~1 in. of Hg.
5. Concentrate.
a. Remove test tubes from vacuum manifold.
b. Evaporate to dryness at 600C using a stream of air or nitrogen gas.
6. Sample Cleanup.
a. Add 200μL of dimethylformamide.
b. Add 1 mL of hexane saturated with dimethylformamide.
c. Mix by inversion for 5 min.
d. Centrifuge at 1500g for 5 min.
e. Transfer lower dimethylformamide layer to a clean test tube.
7. Concentrate.
a. Evaporate to dryness at 500 C using a stream of air or nitrogen gas.
8. Derivatize.
a. Add 100μL of ethyl acetate and 100μL of BSTFA (with 1 % TMCS).
b. Mix/vortex.
c. No heating is required.
9. Quantitate.
a. Inject a 1-to 2 μL sample onto GCMS.
b. Monitor the following ions:
GHB-diTMS 233, 234, 235
GHB-D6-diTMS 239, 240, 241
See Figure 3.
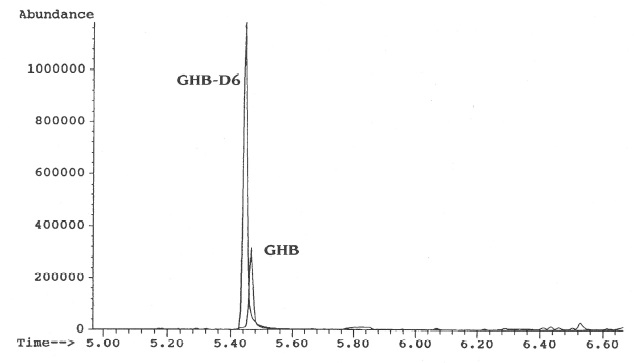
Figure 3: Chromatogram of the selected target ions showing GHB and GHB-D6 from a 5 m g/L extracted urine standard.
Five 50 mg/L standards were prepared by adding 10μg of GHB to 200μL of drug-free urine. These standards were extracted using the previously described procedure. The internal standard (4μg of GHB-D6) was added immediately prior to the evaporation of the DMF. Five 50 mg/L unextracted standards were prepared by adding 10μg of GHB and 4μg of GHB-D6 to a test tube that was dried at 50oC. All samples were derivatized using the procedure previously described. The recovery was calculated by comparing the area under the curve of the target ion for the extracted and the average of the unextracted standards. The average recovery was 67.2% (range =76%-58%).
The assay is linear from 1 to 100 mg/L with the upper range limited by the saturation of the detector. The method could be run with a split injection if higher concentrations need to be quantified.
The diTMS derivatives are stable for more than 7 d at room temperature.
The assay is sensitive to 1 mg/L.
The only interference identified in this procedure was from urea. It is derivatized along with GHB to form urea-diTMS. Using the SIM ions listed above eliminates the interference.
This procedure allows for the direct analysis of GHB and eliminates any possibility of forming or extracting γ-butyrolactone (GBL). Conversion to GBL is problematic in forensic analysis where litigation is involved because GHB is a scheduled drug in many US states and GBL is not. This method was designed to identify low levels of GHB in urine and requires only 200μL of sample. Expected concentrations in biological samples may be much higher and therefore a smaller sample size should be used [6]. The method utilizes a novel copolymeric sorbent employing SPE as a filter. A sample cleanup step and silylation followed by GCMS analysis is also incorporated into the method. The derivatization with BSTFA with 1% TMCS is accomplished without a heating step, owing to the high reactivity of the BSTFA to the GHB. The derivative is stable for more than a week at room-temperature conditions.
SPE-Selective Adsorption
A second approach to SPE is to selectively bond the analyte of interest while allowing the matrix to pass through. This is probably one of the more commonapproaches to SPE. Figure 4 is a graphic demonstration of this approach. The analysis of gabapentin in human serum is presented to illustrate how this sample preparation is performed.
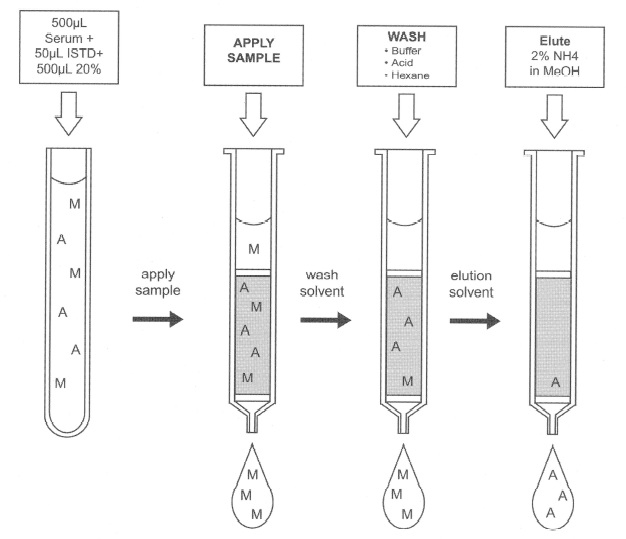
Figure 4: SPE as a selective adsorption tool for gabapentin serum assay.
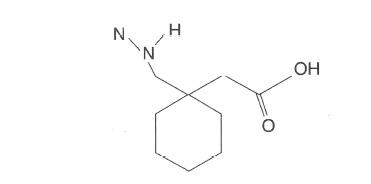
Figure 5: Gabapentin/Neurontin@ [(l-aminomethyl-l-cyclohexyl) acetic acid.
Gabapentin [(l-aminomethyl-l-cyclohexyl) acetic acid] is an anticonvulsant agent used as an adjunctive therapeutic agent in the treatment of seizure disorders not adequately controlled by standard monoanticonvulsant therapy. Adequate drug levels are critical to controlling seizures.Chemically, gabapentin is an interesting drug to isolate because of three functional groups on its molecule an amine group (cationic), a carboxylic acid (anionic), and the ring structure (neutral, reversed phase extraction).
Figure 5 shows the chemical structures of gabapentin and the compound used as the internal standard in this analysis. Section 2.1 shows the extraction of gabapentin from serum or whole blood. Section 2.2 shows the data from the assay validation for this method.
Presented in this application is a specific and selective gas chromatographic method using a nitrogen-phosphorus detector (1). The selectivity of this method is illustrated in Table 2, which lists compounds found not to interfere with the gabapentin assay from more than 40 drugs screened for potential interference with this method. Figure 6 shows the chromatograms of the serum extracts.
*Abbreviations: BUN, blood urea nitrogen; HCG, human chorionic gonadotropin; LDH, lactate dehydrogenase; NAPA, n-acetylprocainamide
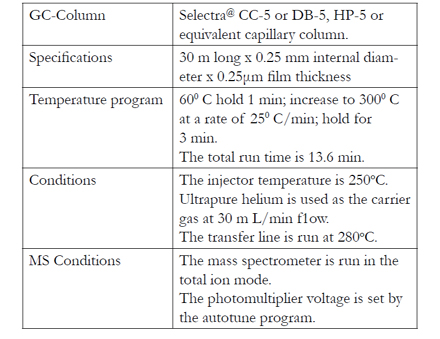
Table 2: Compounds Found Not to Interfere with Gabapentin Assay.
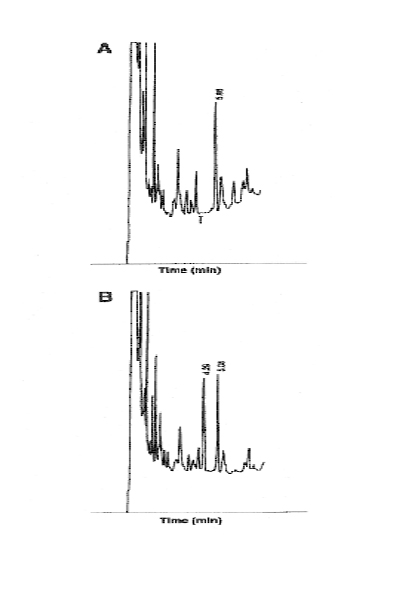
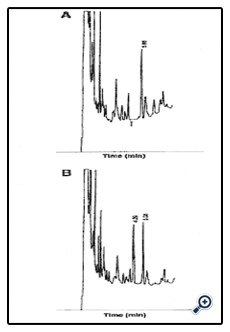
Figure 6: Gabapentin analysis. Chromatograms of serum extracts: (A) Internal standard present (5.06 min). (B) Gabapentin 5.0 m g/L (4.3 min) and internal standard (5.08) present. Time (min).
1. Prepare sample
a. Place 500μL of sample, calibrator, or control into a 10 x 25 mm disposable glass test tube and add 25μL of internal standard (5.0 mg/L).
b. Vortex tube.
c. Add 500μL of 20% acetic acid and vortex tube again.
2. Condition SPE column.
a. 1 x 3 mL of CH3OH; aspirate.
b. 1 x 3 mL of DI H2O; aspirate.
c. 1 x 3 mL of 1 N HC1; aspirate.
3. Apply sample.
a. Load at 1 m L/min.
4. Wash column.
a. 1 x 3 mL of DI H2O; aspirate.
b. 1 x 3 mL of ethyl acetate.
c. 1 x 3 mL of hexane.
d. Dry column (5 min at >10 in. Hg or until column is dry).
5. Elute Gabapentin.
a. 1 x 1 mL of 2% NH4OH in MeOH.
b. Evaporate to dryness at 400C in a water bath.
6. Dervizatize.
a. Add 50μL of MTBSTF A + 1 % BDMCS reagent to the residue.
b. Cap tube and put into a water bath at 700C for 30 min.
c. Remove and allow to cool for 5-10 min.
7. Quantitate.
a. Insert 1-2μL of the sample onto the chromatograph.
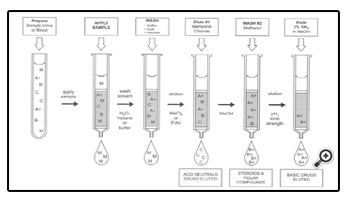
Figure 7: Copolymeric interaction, forensic drug assay.
a. Linearity was observed from 0.2 to 30 mg/L.
b. Within-run precision:
7.0% for 1.1 mg/L (N= 10)
3.4% for 4.5 mg/L (N= 10)
c. Between-run precision:
12% for 1.1 mg/L (N= 16) over 3 wk
5.0% for 4.5 mg/L (N=16) over 3 wk
d. Recovery of gabapentin: Internal standard
1.0 mg/L: 46% 69% (N= 10)
5.0 mg/L: 57%
20.0 mg/L: 51 %
A simple, rapid, and selective method for gabapentin is presented that uses a reversed phase separation. Sample chromatograms can be seen in Figure 6. The pH of the sample was used to suppress the ionization of the carboxylic acid group. Ethyl acetate and hexane were used to wash the matrix away from the sorbentisolated drug.
Gabapentin has a limited solubility in these solvents. Elution at an alkaline pH ionized the acid portion of the drug thus freeing the drug from the sorbent. Increasing the ammonium hydroxide concentration did not improve the recovery of the analyte and could produce ammonium salts, requiring further sample purification.
The derivatization of gabapentin with MTBSTFA and 1 % BDMCS improved the peak shape of the analytes and also increased the molecular weights, thus increasing their retention time on the chromatographic system.
Derivatized gabapentin samples were stable for up to 1 d with a< 10% loss due to chemical degradation.
SPE in Copolymeric Interaction
Figure 7 shows a third approach to SPE, referred to as “copolymeric” or selective extraction. This is a most useful approach in doing class extractions or drugplus- metabolite extractions in groups of compounds. An example of this type of approach can be seen in the general method used for forensic samples.
The versatility of SPE can be best exhibited by its usage in the separation of a wide variety of drugs using a combination of separation strategies. The widest variety of separations have been developed using the combination of a C8 with a cationic exchanger, usually benzene sulfonic acid. CLEAN SCREEN DAU, Bond Elute Certify@, SPECII@, Isolute@, and NARCII@ are a few of the products that currently use this combination and are termed “copolymeric sorbents”.
This mechanism works by providing separation of the acid/neutral drugs using reversed phase C8 functionality; the benzene sulfonic acid mechanism works on basic drugs by cation exchange of the amine functionalities. A variety of drugs that have been extracted by the CLEAN SCREEN DAU extraction column using the method found in sections 3.1 and 3.2. This method is an example of the kinds of methods that are developed in toxicology laboratories where a general method is used to identify drugs in biological samples for clinical management and toxicological significance. Section 3.2 shows the extraction procedure used for forensic drug analysis.
Selectra@ is a registered trademark of United Chemical Technologies, Inc. (Bristol, PA). HP 5@ is a registered trademark of Hewlett Packard Corporation (Palo Alto, CA). DB-5@ is a registered trademark of J & W Scientific Inc. (Folsom, CA). Bond Elute Certify@ is a registered trademark of Varian, Inc. (Harbor City, CA). SPECII@ is a registered trademark of Ansys (Harbor City, CA). Isolute@ is a registered trademark of International Sorbent Technologies, Inc. (Cardiff, UK). NARCII@ is a registered trademark of J. T. Baker (Phillipsburg, NJ).
1. Prepare sample.
1.1 Urine.
a. To 5 mL of urine add 50-300μL of 1.0 M acetic acid to adjust sample pH to between 4.8 and 5.5.
1.2 Whole blood:
a. To 2 mL of blood add 8 mL of DI H2O. Mix/vortex and let stand 5 min.
b. Add 150-300μL of 1.0 M acetic acid to adjust sample pH to between 4.8 and 5.5.
c. Centrifuge for 10 min at 670g and discard pellet.
1.3 Tissue:
a. Homogenize 1 part tissue with 3 parts of DI H2O..
b. Centrifuge for 10 min at 670g and discard pellet.
c. Transfer 10 mL of supernatant to a clean tube.
d. Add 150-300μL of 1.0 M acetic acid to adjust sample pH to between 4.8 and 5.5.
2 Condition CLEAN SCREEN extraction column.
a. 1 x 3 mL of CH3OH; aspirate.
b. 1 x 3 mL of DI H2O; aspirate.
c. 1 x 1 mL of 0.1 M acetic acid; aspirate.
Note: Aspirate at ≦3 in. Hg to prevent sorbent drying.
3 Apply sample.
a. Load at 1-2 mL/ min.
4 Wash column.
a. 1 x 3 mL of 0.1 M phosphate buffer, pH 6.0;aspirate.
b. 1 x 1 mL of 0.1M acetic acid; aspirate.
c. Dry column (5 min at ≦10 in. Hg).
d. 1 x 3 mL of hexane; aspirate.
5 Elute acidic and neutral drugs (faction A).
a. 2 x 2 mL of CH2Cl2; collect eluate at ≦5 mL/min.
b. Evaporate to dryness at ≦40oC.
6 Extract and analyze faction A.
a. Add 1 mL of hexane and 1 mL of CH3OH/H2O (80:20). Mix/vortex.
b. Centrifuge to separate layers. Aspirate and discard hexane (upper) layer.
c. Evaporate again to dryness at ≦40oC.
d. Reconstitute with 100μL of ethyl acetate and inject 1 to 2μL onto chromatograph.
7 Wash column.
a. 1 x 2 mL of methanol; aspirate.
b. Dry column (5 min ≦10 in. Hg).
8 Elute basic drugs (fraction B).
a. 1 x 2.0 mL of methanol-NH4OH (98:2); collect eluate at 1-2 m L/ min.
Note: Prepare elution solvent daily.
9 Extract and analyze fraction B.
a. Add 3.0 mL of DI H2O and 250 μL chloroform to eluate. Mix/vortex 30 s.
b. Centrifuge to separate phases. Aspirate and discard aqueous (upper) layer.
c. Inject 1-2μL of the chloroform layer onto Hideharu Shintani, International Journal of Clinical Pharmacology & Toxicology 2013, 2:602 10 chromatograph. Note: Fractions A and B can be combined before analysis and evaporated together.
The future of SPE is illustrated in Fig. 8, which shows that immunoaffinity can be used to isolate the analytes of interest. This type of SPE is dependent on the attachment of a biologically active molecule to a matrix surface by the use of an aldehydic silane. This forms a covalent bond with the amine groups of the substrate, allowing for the immobilization of the biomolecule with the surface of a variety of materials.
SPE is a very robust technique; however, what if we could improve on the selectivity of our separations by the use of antibodies, enzymes, peptides, proteins, or the attachment of any biological substrate to pick up our analytes?
This type of technology was introduced many years ago by the gluteraldehyde procedure [1-3] and was used for the immobilization of antibodies for benzodiazepines [4]. This immunoaffinity column could pick up most benzodiazepines including their glucuronides. This process could eliminate the acid-base or enzymatic hydrolysis step in conventional SPE procedures.
The only problems with this procedure were in the formation of two Schiff bases in the covalent linkage, making it sterically strained and also susceptible to hydrolysis by various mechanisms. These two limitations represented challenges that limited the effectiveness of this procedure. The recent introduction of a new line of aldehydic silanes (Bio Conext) offers many advantages above the present technologies in ligand immobilization. Aldehydic silanes attach easily to many matrices that contain hydroxyl functions. This includes glass, ELISA plates, silica, plastic beads, metal, agarose, and many polymeric resins. The bonding to primary amines of the substrate creates only one Schiff base attachment. This provides for a more stable linkage that is not sterically hindered. The use of various carbon lengths in the chains from the matrix surface to the biomolecule allow access to active sites and also allow the binding to larger size molecules.
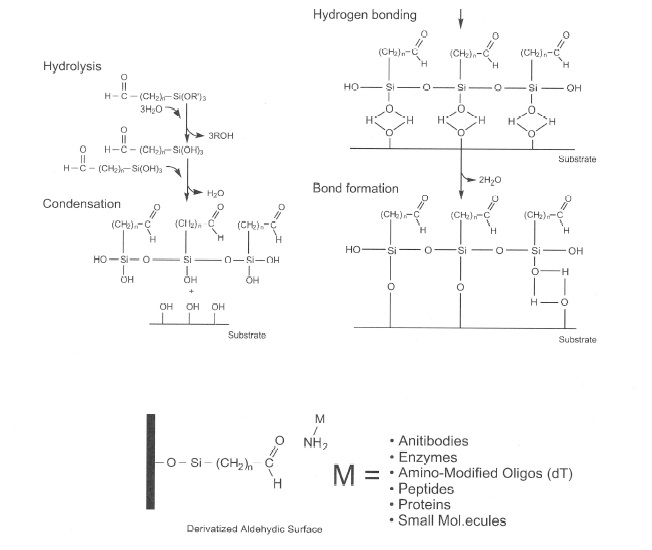
Figure 8: Aldehydic silane attachment.
Figure 8 illustrates the chemistry involved in the attachment of the aldehydic silane to a biologically active substrate. Figure 9 explains, in a step-by-step fashion, an actual synthesis of bonding the aldehydic silane first to the matrix (silica) then to Protein A. Figure 10 illustrates how an immunoaffinity procedure would be performed. The simplicity of the method, along with selectivity and the limitless possibilities for attachment and analysis, illustrate the direction that SPE in the future may take.
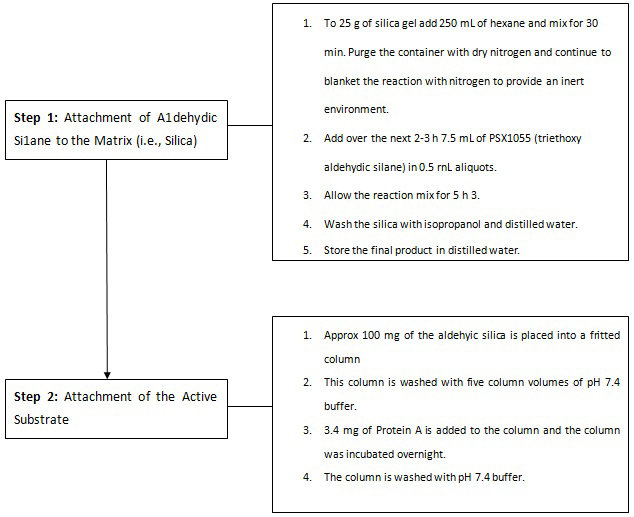
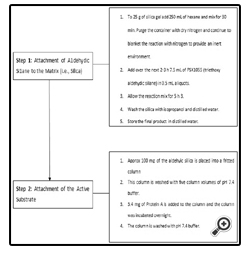
Figure 9: Aldehydic silane bonding.


Figure 10: Immunoaffinity SPE Procedure.
References
- Dyer, J. Gamma-hydroxybutyrate: A health-food product producing coma and seizure-like activity. Am. J. Emerg. Med. 9:321-324, 1991.
- Chin, M. Acute poisoning from gamma-hydroxybutyrate in California.Wes. J. Med. 156:380-384, 1992.
- Andrews, K. Getting the scoop on gamma-hydroxybutyrate or GHB: The new recreational drug. Proceedings of the 49th Annual Meeting of the American Academy of Forensic Sciences, Historical Overview, p. 3, 1997.
- Dean, W., Morgenthaler, J., and Fowkes, S. W. GHB: The Natural Mood Enhancer. Smart Publications, 1997.
- Gibson, K. Stable isotope dilution analysis of 4-hydroxybutyric acid: An accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. Biomed. Environ. Mass Spectrom. 19:89-93, 1990.
- Stephens, B. and Baselt, R. Driving under the influence of GHB. J. Analyt. Toxicol. 18:357.358, 1994.
- Wolf 11, C. E., Sady, J., and Pokalis, A. Determination of gabapentin in serum using solid phase extraction and gas-liquid Chromatography. J. Analyt. Toxicol. 20:498-501, 1996.
- Neurath, A., and Strick, N. Enzyme linked fluorescent immunoassay using beta-galactosidase and antibodies covalentyly bound to polystyrene plates. J. Virol. Methods 3:155-165, 1981.
- Weston, P., and Avrameas, S. Proteins coupled to polyacrylamide beads using glutaraldehyde Biochem. Biophys. Re. Commun, 45:1574-1580, 1971
- Terynck, T., and Avrameas, S. Polyacrylamide-protein immunoadsorbents prepared with gluteraldehyde. FEBS Lett. 23:24-28, 1972.
- Franzelius, C., Ackermann, I., Deinl, I., Angermaier, L., and Machbert, G. Simultaneous extraction of selected benzodiazepines and benzodiazepine glucuronides from urine by immunoadsorption. J. Analy. Toxicol. 22: 359-362, 1998.