T-Helper Cell Cytokine Expression Profiling in Rheumatoid Arthritis Patients by Flow Cytometric Bead Array Analysis
Chen J1, Yang J1, Ding L1, Shaoliang Jie1, Rongzhong Huang3, Zhang SR1, Zili Fu2*
1 Department of Rheumatology, the Second Hospital of Shanxi Medical University, Taiyuan, China.
2 Department of Rheumatology, the First Hospital of Shanxi Medical University, Shanxi, China.
3 Department of Rehabilitation Medicine, The second Affiliated Hospital of Chongqing Medical University, Chongqing, P. R.China.
*Corresponding Author
Zili Fu,
Professor, Department of Rheumatology,
The First Hospital of Shanxi Medical University,
No. 85 South Jiefang Road, Yingze District, Shanxi 030001, China.
Tel: +86-0351-4639526
Fax: +86-0351-4639010
E-mail: fuzili1234@sina.cn
Received: June 28, 2015; Accepted: July 28, 2015; Published: August 04, 2015
Citation: Zili Fu et al., (2015) T-Helper Cell Cytokine Expression Profiling in Rheumatoid Arthritis Patients by Flow Cytometric Bead Array Analysis. Int J Clin Ther Diagn. S2:001, 1-5. doi: dx.doi.org/10.19070/2332-2926-SI02001
Copyright: Zili Fu© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Rheumatoid arthritis (RA) is the most common chronic autoimmune disease affecting multiple joints. A
chronic imbalance in cytokine production by T-helper (Th) cells is likely a key factor in RA development. Our objective was to profile the serum cytokine expression from three key Th cell types (Th1, Th2, and Th17) in RA patients in order to correlate the resulting cytokine expression profiles with RA activity.
Material and Methods: From a population of RA patients (n = 71) and healthy controls (n = 18), the serum concentrations of seven cytokines (IL-2, IL-4, IL-6, IL-10, IL-17A, IFN-γ, and TNF-α) were analyzed by flow cytometric bead array (CBA).
Results: The serum concentrations of all seven cytokines were significantly higher in RA patients than in healthy controls. Interestingly, the serum concentration profiles varied with the 28-joint Disease Activity Score (DAS28), a measure of RA activity derived from joint indices (tender joints and swollen joints count) and the erythrocyte sedimentation rate. In the high RA activity group (DAS28 > 5.1), all seven cytokines were significantly elevated. In the moderate RA activity group (DAS28 between 3.2 and 5.1), only IL-2, IL-6, IL-10, and IL-17A were significantly increased. In the low RA activity group (DAS28 ≤ 3.2), only IL-2, IL-4, and TNF-α were significantly elevated.
Conclusions: The Th cell-derived cytokine expression profile significantly changes across varying levels of RA activity. Th1/Th17 cell-derived TNF-α and Th2 cell-derived IL-4 appear to play more important roles in the early stages of RA, while all seven cytokines derived from Th1, Th2, and Th17 cells (IL-2, IL-4, IL-6, IL-10, IL-17A, IFN-γ, and TNF-α) are overtly involved in the advanced stages of RA.
2.Introduction
3.Materials and Methods
3.1 Ethics Statement
3.2 Subject Recruitment & Sample Collection
3.3 CBA Standards Preparation
3.4 CBA of Cytokine Expression
3.5 Statistical Analysis
4.Results
5.Discussion
6.Conclusion
7.Acknowledgments
8.Author Contributions
9.References
Keywords
Rheumatoid Arthritis: RA; T-helper: Th1; Th2; Th17; Cytokine; Cytometric Bead Array; CBA.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease affecting approximately 1% of the population in the Western world characterized by chronic inflammation of the joint synovium by activated inflammatory leukocytes (such as T-helper [Th] lymphocytes and monocytes), synovial hyperplasia, neo-angiogenesis, and progressive destruction of cartilage and bone [1]. The pathogenesis of RA is a multi-factorial process involving genetic and environmental factors that can result in systemic immunological dysfunction under certain conditions [2]. Specifically, inappropriate regulation of CD4+ Th cells has been implicated in the pathophysiology of both RA and systemic lupus erythematosus (SLE) [3, 4], and several studies have shown that RA is associated with Th1/Th17 cell cytokine imbalances [5, 6]. As cytokine-driven activation of CD4+ Th cells is critical to the elimination of pathogens, a chronic imbalance in cytokine production by Th cells is likely a key factor in the development of RA.
Therefore, in this work, we profiled the cytokine expression from three key Th cell types (Th1, Th2, and Th17) in peripheral blood samples from RA patients by flow cytometric bead array (CBA) in order to correlate the resulting cytokine expression profiles with RA activity.
Materials and Methods
The study protocol was approved by the Regional Committee on Ethics for Human Research at the Faculty of the Second Hospital of Shanxi Medical University (Taiyuan, China). Prior to participation in the study, written informed consent was obtained from all subjects after a full explanation of the procedures.
The study population consisting of 71 RA patients (21 males and 50 females aged 18-75 years, mean age: 51.20 ± 12.18 years) were included based on the revised classification criteria of the American College of Rheumatology [7] (Table 1). All patients were seen at the outpatient immunology clinic at our hospital between March 2009 and February 2010. Disease duration ranged from 6 months to 13 years (mean duration: 5.24 ± 0.77 years). RA activity was assessed by the 28-joint Disease Activity Score (DAS28), which is derived from joint indices (tender joints and swollen joints count) and the erythrocyte sedimentation rate (ESR) [7]. From the total study population of 71 RA patients, 14 patients were segregated into a low RA activity group (DAS28 ≤ 3.2), 22 patients were segregated into a moderate RA activity group (3.2 < DAS28 ≤ 5.1), and 35 patients were segregated into a high RA activity group (DAS28 > 5.1). Eighteen healthy controls (5 males and 13 females aged 22–54 years, mean age: 47.48 ± 14.43) were also enrolled from our hospital during the same time period.
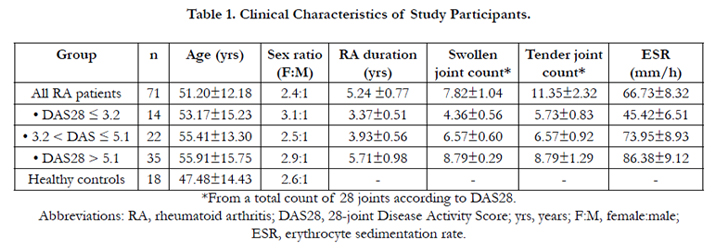

Table 1. Clinical Characteristics of Study Participants.
*From a total count of 28 joints according to DAS28.
Abbreviations: RA, rheumatoid arthritis; DAS28, 28-joint Disease Activity Score; yrs, years; F:M, female:male;
ESR, erythrocyte sedimentation rate.
After consenting, 5.0ml of blood was collected from each of the 76 RA patients and 18 healthy controls at the time of clinical assessment. Serum was separated from the specimens at 1000 rpm for 15 min at 20°C. After centrifugation, sera were collected and stored at −80°C for later analysis.
Preparation of the CBA standards was performed according to the kit instructions (BD CBA Cell Signaling Flex Set, Becton Dickinson, USA). Briefly, Standard Tubes (12 × 75 mm) were labeled and arranged in the following order for purposes of serial dilution: Top Standard, 1:2, 1:4, 1:8, 1:16, 1:32, 1:64, 1:128, and 1:256. Then, 20μl of each Cell Signaling BD CBA Flex Set Standard was added to the Top Standard tube. The Assay Diluent was added into the Top Standard tube to bring the final volume to 1 ml. Then, 500μl of Assay Diluent was added into each of the remaining tubes. Serial dilution was performed by transferring 500 μl from the Top Standard tube to the 1:2 dilution tube, mixing thoroughly, and then repeating this process for the remaining tubes. The Assay Diluent served as the negative control.
The 50μl standard dilutions (prepared above) and test samples were added to the appropriate tubes. Then, 50μl mixed Capture Beads were transferred into each assay tube. The assay tubes were incubated for three hours at room temperature. Then, 50μl mixed PE-conjugated Detection Reagent was added to each assay tube and then incubated at room temperature for one hour in a dark room. The assay samples were washed with 1.0ml Wash Buffer and centrifuged at 1500 rpm for 5 minutes. Then, 300μl Wash Buffer were transferred into each assay tube.
The assay samples were analyzed by CBA (BD FACS Calibur Flow Cytometer and FCAP Array™ software). Seven cytokines were detected: interleukin-2 (IL-2), interleukin-4 (IL-4), interleukin-6 (IL-6), interleukin-10 (IL-10), tumor necrosis factor (TNF-α), interferon-γ (IFN-γ), and interleukin-17A (IL-17A) (Figure 1). Because each cytokine was coated with a uniquely-sized microsphere, each cytokine could be easily distinguished by CBA. The concentration of each cytokine was determined according to its fluorescence intensity (Figure 1).
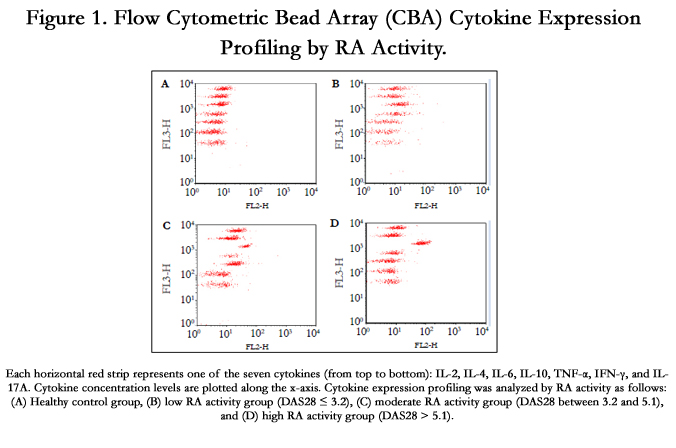
Figure 1. Flow Cytometric Bead Array (CBA) Cytokine Expression Profiling by RA Activity.
Each horizontal red strip represents one of the seven cytokines (from top to bottom): IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ, and IL- 17A. Cytokine concentration levels are plotted along the x-axis. Cytokine expression profiling was analyzed by RA activity as follows: (A) Healthy control group, (B) low RA activity group (DAS28 ≤ 3.2), (C) moderate RA activity group (DAS28 between 3.2 and 5.1), and (D) high RA activity group (DAS28 > 5.1).
Data were analyzed using SPSS 13.0 (IBM, USA). The experimental groups were compared by one-way analysis of variance (ANOVA) and Tukey's method. Student’s t-test was used to compare the RA and healthy control groups. P < 0.05 was considered statistically significant for all analysis.
Results
CBA, which discriminates between microparticles on the basis of size and fluorescence by flow cytometry, takes significantly less time than Western blotting and provides quantitative results with a wider dynamic range than conventional ELISA. Here, seven Th cell-derived cytokines in the serum of RA patients and healthy controls were measured using CBA (Figure 1). As a result, the serum concentrations of IL-2, IL-4, IL-6, IL-10, IL-17A, IFN-γ, and TNF-α were significantly higher than those of healthy controls (p < 0.001; Table 2). However, the serum concentration profiles of these various cytokines significantly varied with DSA28- based RA activity (Table 3). Specifically, in the high RA activity group (DAS28 > 5.1), the serum concentrations of IL-2, IL-4, IL- 6, IL-10, IL-17A, IFN-γ, and TNF-α were significantly elevated. In the moderate RA activity group (DAS28 between 3.2 and 5.1), only the serum concentrations of IL-2, IL-6, IL-10, and IL-17A were significantly increased. In the low RA activity group (DAS28 ≤ 3.2), only the serum concentrations of IL-2, IL-4, and TNF-α were significantly elevated.
Discussion
Cytokines are a group of small molecules secreted by Th cells (and non-immune cells as well) that play an important role in the pathogenesis of RA by promoting autoimmune and long-term inflammatory synovitis that results in neighboring joint tissue destruction [8]. The different types of Th cells secrete an array of cytokines at varying levels of expression in response to multiple stimuli, which results in a complex network of self-regulating interrelationships [9]. Imbalances in this Th cell-cytokine network likely play an important role in the chronic inflammation and joint damage underlying RA [10].
Th1 cells primarily secrete IFN-γ, IL-2, and TNF-α. IFN-γ is a key macrophage activating factor that enhances their lethality and also stimulates NK cells [11]. IL-2 is important in the development of CD4+ T-memory cells and is a key factor in CD8+ T-cell stimulation. TNF-α, which is also secreted by mononuclear macrophages and Th17 cells, enhances expression of endothelial cell adhesion molecules (such as ICAM-1), promotes the adhesion of leukocytes to the endothelium, and enhances vascular endothelial permeation. TNF-α also promotes synovial cells, macrophages, fibroblasts, and chondrocytes to produce IL-1, IL-8, and TNF-α [12]. TNF-α is also produced by activated synoviocytes within the inflamed intimal lining layer of the synovial tissue in RA patients. Moreover, TNF-α is also expressed by activated T cells following estrogen loss and promotes bone-resorbing osteoclast (OCL) formation by upregulating osteoblast production of RANKL and augmenting OCL precursor responses to RANKL [13]. Because of TNF-α’s key role in promoting synovial inflammation and bone loss, anti-TNF-α therapy is now a well-established therapeutic strategy for RA patients and has been shown to act synergistically with methotrexate to improve RA symptoms [14].
Th2 cells produce IL-4 and IL-10. IL-4 prompts the differentiation of Th2 cells, forming a positive feedback loop for Th2 cell differentiation [4]. IL-10 inhibits the proliferation of Th1 cells and impairs dendritic cell function [15]. Th2 cells also restrain inflammatory reactions by inhibiting Th1 and antigen-present cells.
Th17 cells produce TNF-α, IL-6, and IL-17A. Along with TNF-α, IL-6 is also a major inflammatory mediator in RA. Although IL-6 does not directly stimulate synovial mother cells and chondrocytes to produce PGE2 and collagenase, it does enhance the effects of TNF-α and IL-1 as well as affecting IL-1, IL-2, and TNF-α production [16]. IL-17A induces inflammation and neutrophil activation and has been found to induce bone and cartilage destruction in RA patients [17, 18].
In the current study, serum concentrations of IL-2, IL-4, IL-6, IL-10, IL-17A, IFN-γ, and TNF-α were determined to be significantly higher in RA patients relative to healthy controls (Table 2). As these particular cytokines as primarily secreted by Th1, Th2, and Th17 cells, this study suggests that these Th cells actively participate the pathogenesis of RA. This conclusion is consistent with that of our previous study [19]. Moreover, we also profiled the expression of these seven cytokines by RA activity. In RA patients with high DAS28 scores, all seven cytokines were significantly higher as compared to healthy controls (Table 3). However, in RA patients with moderate DAS28 scores, the serum concentrations of IL-2, IL-6, IL-10, and IL-17A were significantly elevated, but those of IL-4, IFN-γ, and TNF-α were not significantly elevated, compared to healthy controls. Moreover, in RA patients with low DAS28 scores, the serum concentrations of IL-2, IL-4, and TNF-α were significantly elevated, but those of IL-6, IL-10 and IL-17A were not significantly elevated, compared to healthy controls.
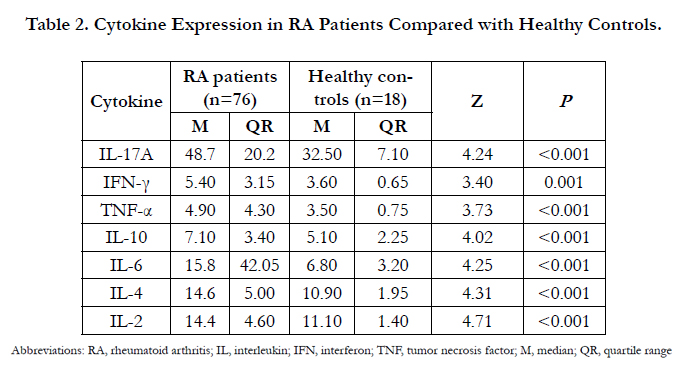
Table 2. Cytokine Expression in RA Patients Compared with Healthy Controls.
Abbreviations: RA, rheumatoid arthritis; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; M, median; QR, quartile range
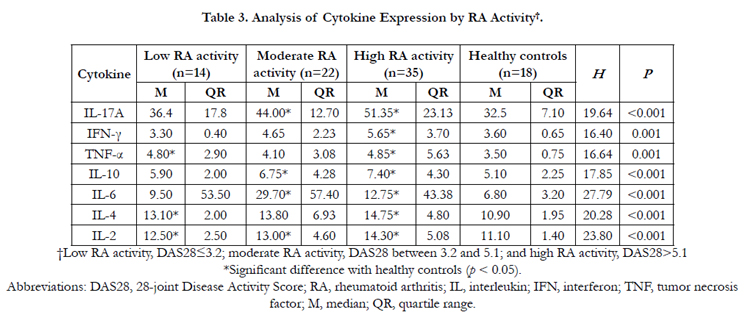
Table 3. Analysis of Cytokine Expression by RA Activity†.
†Low RA activity, DAS28≤3.2; moderate RA activity, DAS28 between 3.2 and 5.1; and high RA activity, DAS28>5.1 *Significant difference with healthy controls (p < 0.05).
Abbreviations: DAS28, 28-joint Disease Activity Score; RA, rheumatoid arthritis; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; M, median; QR, quartile range.
Therefore, this study reveals that these seven cytokines are differentially expressed during according to varying levels of RA activity. For example, Th1 cell-derived IL-2 derived was consistently and significantly increased across all levels of RA activity. In contrast, IL-4 and TNF-α were significantly elevated in the low RA activity group but not in the moderate RA activity group. Based on these findings and our current understanding of RA, Th1, Th2, and Th17 cells appear to play different but collaborative roles during the development of RA [15]. Th1 and Th17 cells appear to trigger early-phase inflammation through TNF-α secretion, and then Th17 cells appear to further aggravate inflammation in the sustained phase of RA through secretion of IL-17A and IL-6. Finally, Th1 cells appear to promote late-phase inflammation through IFN-γ and IL-2 secretion. Consistent with this viewpoint, a previous study on experimental autoimmune encephalomyelitis (EAE) mice – an animal model of multiple sclerosis (MS) -- showed that absence of the transcription factor T-bet (which is essential for the development of Th1 cells) protected mice from EAE [20] Similar to RA, EAE and MS are autoimmune disease states mediated by CD4+ cells in which Th1 cells play an important role, suggesting that Th1 cells play pathogenic roles in all these autoimmune conditions. Another study on EAE mice found that the expression of Th17-derived cytokines occurred earlier than that of Th2 cell-derived cytokines and that Th2 cell-derived IFN-γ expression remained elevated long after Th-17 cell-derived IL-17A expression had subsided [21].
Moreover, Th2 cells appear to switch from IL-4 to IL-10 production as RA progresses. Accordingly, synovial fluid samples from patients with very early RA possess elevated IL-4 levels, whereas synovial fluid samples from patients with established RA lacks IL- 4. Moreover, IL-4 inhibits OCL formation through prompting osteoblasts to produce osteoprotegerin [22, 23], and local IL-4 overexpression has been shown to forestall joint damage and bone erosion while suppressing messenger RNA levels of IL-17, IL-12, and cathepsin K as well as promoting IL-6 and IL-12 protein production in a collagen arthritis mouse model; moreover, IL-4 has been shown to suppress Type I collagen breakdown and promote type I procollagen synthesis in bone samples of arthritis patients in vitro [24]. As IL-4 displays a protective effect on the synovium and bone, these combined findings suggest that Th2 cells switching from IL-4 to IL-10 production may be a contributing factor in the development of more severe forms of RA.
Conclusion
This study reveals that the Th cell-derived cytokine expression profile significantly changes across varying levels of RA activity. Th1/Th17 cell-derived TNF-α and Th2 cell-derived IL-4 appear to play more important roles in the early stages of RA, while all seven cytokines derived from Th1, Th2, and Th17 cells (IL-2, IL- 4, IL-6, IL-10, IL-17A, IFN-γ, and TNF-α) are overtly involved in the advanced stages of RA. Larger-scale, multicenter studies performing CBA-based cytokine expression profiling on more demographically heterogeneous RA patient populations (including subgroups for environmental confounders of cytokine expression such as smoking status and elevated BMI levels) are needed to validate these promising findings.
Acknowledgments
All authors thank the patients who took part in the study and the staff in our department who helped with patient recruitment. This research greatly benefited from discussions with Professor Xiaofeng Li and Dr. Jingchao Liu. We also thank Professors Zhenming Pei and Guoqing Ji for their critical reviews of the manuscript. This work was supported by the Project for Scientific and Technological Social Development of Shanxi Province, China (grant no. 20120313025-4).
Author Contributions
Conceived and designed the experiments: Junwei Chen. Performed the experiments: Shaoran Zhang, Chenglan Yan, and Shaoliang Jie. Analyzed the data: Lijuan Ding and Junwei Chen. Contributed reagents/materials/analysis tools: Jianfang Xie and Meng Wu. Wrote the paper: Jinhua Yang and Zili Fu.
References
- Abdel-Nasser AM, Rasker JJ, Vaikenburg HA (1997) Epidemiological and clinical aspects relating to the variability of rheumatoid arthritis. Semin Arthritis Rheum 27(2): 123-140.
- Tobón GJ, Youinou P, Saraux A (2010) The environment, geo-epidemiology, and autoimmune disease: rheumatoid arthritis. J Autoimmun 35(1): 10-14.
- Goodnow CC, Sprent J, de St Groth BF, Vinuesa CG (2005) Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 435(7042): 590-597.
- McInnes IB, Liew FY, Gracie JA (2005) Interleukin-18: a therapeutic target in rheumatoid arthritis. Arthritis Res Ther 7(1): 385-41.
- Veys EM, Menkes CJ, Emery P (1997) A randomized, double‐blind study comparing twenty‐four‐week treatment with recombinant interferon‐γ versus placebo in the treatment of rheumatoid arthritis. Arthritis Rheum 40(1): 62-68.
- Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ (2000) Clonality and longevity of CD4+ CD28null T cells are associated with defects in apoptotic pathways. J Immunol 165(11): 6301-6307.
- Prevoo ML, Van't Hof MA, Kuper HH, Van Leeuwen MA, Van de Putte LB, et al. (1995) Modified disease activity scores that include twenty‐eightjoint counts development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 38(1): 44-48.
- Andreakos ET, Foxwell BM, Brennan FM, Maini RN, Feldmann M (2002) Cytokines and anti-cytokine biologicals in autoimmunity: present and future. Cytokine Growth Factor Rev 13(4-5): 299-313.
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, et al. (2013) Dynamic regulatory network controlling TH17 cell differentiation. Nature 496(7446): 461-468.
- Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF (2013) How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med 19(7): 822-824.
- Palmer G, Mezin F, Juge-Aubry C, Plater-Zyberk C, Gabay C, et al. (2004) Interferon β stimulates interleukin 1 receptor antagonist production in human articular chondrocytes and synovial fibroblasts. Ann Rheum Dis 63(1):43-49.
- Dayer JM (2002) Interleukin 1 or tumor necrosis factor-alpha: which is the real target in rheumatoid arthritis? J Rheumatol Suppl 65: 10-15.
- Boyce BF, Li P, Yao Z, Zhang Q, Badell IR, et al. (2005) TNF-alpha and pathologic bone resorption. Keio J Med 54(3): 127-131.
- Feldmann M, Brennan FM, Foxwell BM, Taylor PC, Williams RO, et al. (2005) Anti-TNF therapy: where have we got to in 2005? J Autoimmun 25: 26-28.
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, et al. (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100(6): 655-669.
- Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, et al. (2003) Anti–interleukin‐6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum 48(6): 1521-1529.
- Feldmann M, Brennan FM, Maini RN (1996) Role of cytokines in rheumatoid arthritis. Ann Rev Immunol 14(1): 397-440.
- Miossec P (2013) IL-17 and Th17 cells in rheumatoid arthritis and other inflammatory conditions. In IL-17, IL-22 and Their Producing Cells: Role in Inflammation and Autoimmunity. Springer Basel 233-242.
- Chen J, Li J, Gao H, Wang C, Luo J, et al. (2012) Comprehensive evaluation of different T-helper cell subsets differentiation and function in rheumatoid arthritis. BioMed Research International 2012.
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, et al. (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200(1): 79-87.
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, et al. (2007) Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 13(4): 423-431.
- Mirosavljevic D, Quinn JM, Elliott J, Horwood NJ, Martin TJ, et al. (2003) T‐Cells Mediate an Inhibitory Effect of Interleukin‐4 on Osteoclastogenesis. J Bone Miner Res 18(6): 984-993.
- Yamada A, Takami M, Kawawa T, Yasuhara R, Zhao B, et al. (2007) Interleukin‐ 4 inhibition of osteoclast differentiation is stronger than that of interleukin‐13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology 120(4): 573-579.
- Lubberts E, Joosten LA, Chabaud M, van Den Bersselaar L, Oppers B, et al. (2000) IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest 105(12): 1697-1710.