A Case of Dandy-Walker Malformation Complicated by Axenfeld-Rieger Syndrome
Ueki M1, Maeda M1, Sugiyama T1,2*, Kohmoto R1, Kojima S1, Ikeda T1, Harada A3, Kanemura Y4, Miya F5,6, Tsunoda T5,6, Yamasaki M3
1 Department of Ophthalmology, Osaka Medical College, Takatsuki, Osaka, Japan.
2 Nakano Eye Clinic of Kyoto Medical Co-operative, Kyoto, Japan.
3 Department of pediatric neurosurgery, Takatsuki General Hospital, Takatsuki, Osaka, Japan.
4 Division of Regenerative Medicine, Institute for Clinical Research, Osaka National Hospital, National Hospital Organization, Osaka, Japan.
5 Department of Medical Science Mathematics, Medical Research Institute, Tokyo Medical and Dental University, Tokyo, Japan.
6 Laboratory for Medical Science Mathematics, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
*Corresponding Author
Tetsuya Sugiyama, MD, PhD,
Department of Ophthalmology, Osaka Medical College, Takatsuki, Osaka, Japan.
Nakano Eye Clinic of Kyoto Medical Co-operative, Kyoto, Japan.
Tel: +81-75-8014151
E-mail: tsugiyama@kyo-con.or.jp
Received: November 01, 2017; Accepted: December 02, 2017; Published: December 04, 2017
Citation: Ueki M, Maeda M, Sugiyama T, Kohmoto R, Kojima S, Ikeda T, et al., A Case of Dandy-Walker Malformation Complicated by Axenfeld-Rieger Syndrome. Int J OpthalmolEye Res, 2017;S1:02:001:1-3. doi: dx.doi.org/10.19070/2332-290X-SI02-01001
Copyright: Khalil NM© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any mediu, provided the original author and source are credited.
Abstract
Purpose: To report a case of Dandy-Walker malformation complicated by Axenfeld-Rieger syndrome.
Case: A 20-day-old female infant underwent ventriculo-peritoneal shunt for hydrocephalus associated with Dandy-Walker malformation when she was 2 days old. She was referred to us because she had corneal opacity and ocular hypertension in both eyes. At the initial examination, her intraocular pressure (IOP) was 35 mmHg in the right eye and 28 mmHg in the left eye, and corneal opacity and extended corneal diameter was present in both eyes. Trabeculotomy was performed twice in both eyes. Her father had also undergone trabeculotomy on both his eyes when he was 3 months old.
Results: Her IOP first stabilized to within the normal limit, but iris atrophy later developed in her left eye. After confirming a mutation in FOXC1, we diagnosed her with Axenfeld-Rieger syndrome. Genetic tests revealed the same mutation in her father.
Conclusion: Since FOXC1ha mutations are associated with Axenfeld-Rieger anomaly as well as Dandy-Walker malformation, these diseases can coexist. Genetic tests may be useful for the diagnosis, treatment, and prognosis determination of patients with congenital glaucoma.
2.Introduction
3.Case Report
4.Discussion
5.Conclusion
6.Acknowledgments & Declarations
7.References
Keywords
Axenfeld-Rieger Syndrome; Dandy-Walker Malformation; FOXC1.
Introduction
One of the developmental anomalies associated with congenital glaucoma is Axenfeld-Rieger Syndrome (ARS). ARS consists of a spectrum of developmental defects of the anterior chamber of the eye, with wide variability of expression. The Axenfeld anomaly is a white line in the posterior aspect of the cornea, near the limbs, and tissue strands extend from the periphery of the iris to this prominent line. The Rieger anomaly involves mesodermal digenesis of the cornea and iris. When these defects are associated with extra ocular findings of maxillary hypoplasia, hypodontia, microdontia, and umbilical abnormalities, the disorder is defined as ARS. This disorder is seen in approximately 1 per 200,000 live births. Its penetrance is complete, although expressivity is variable, and it is associated with a 50% risk of glaucoma. ARS is a genetically heterogeneous group of abnormalities, caused by mutations in at least four different gene loci. Mutations in PITX2 on ch 4q25, FOXC1 on 6p25, PAX6 on 11p13, and FOXO1A on 13q14 have been associated with the development of ARS [1-5].
On the other hand, Dandy-Walker malformation (DWM) is a congenital condition that occurs during embryonic development of the cerebellum and the fourth ventricle [6]. DWM has a frequency of approximately 1 per 25,000-35,000 live births and affects more females than males. Although there are some reports that DWM is caused by heterozygous loss of zinc finger genes (ZIC1 and ZIC4) or mutations affecting FOXC1 and FGF17 [6-11], the etiology is still unclear.
Previously, there have been a few reports regarding patients with a combination of DWM and Axenfeld-Rieger anomaly or glaucoma (but not ARS) [11, 12]. In this report, we present a rare case of a patient with ARS complicated with DWM, whose father was also diagnosed with ARS.
Case Report
A 20-day-old female infant was diagnosed with DWM with enlargement of the fourth ventricle, severe cerebella atrophy, and posterior fossa cyst. When she was delivered by caesarean section at 35 weeks of gestation, corneal opacity was found in both her eyes (Figure 1). She underwent an ophthalmic examination on Day 6 after birth. Both her corneas were cloudy and intraocular pressure (IOP) was 32mmHg in her right eye (OD) and 25mmHg in the left eye (OS) by icare® (ICARE FINLAND OY, Vantaa, Finland). Since she had been diagnosed with congenital glaucoma, treatment with anti-glaucoma eye drops was initiated. However, treatment with dorzolamide (Trusopt®) and latanoprost (Xalatan®) for about 2 weeks were not sufficiently effective. Therefore, she was referred to us for glaucoma surgery.
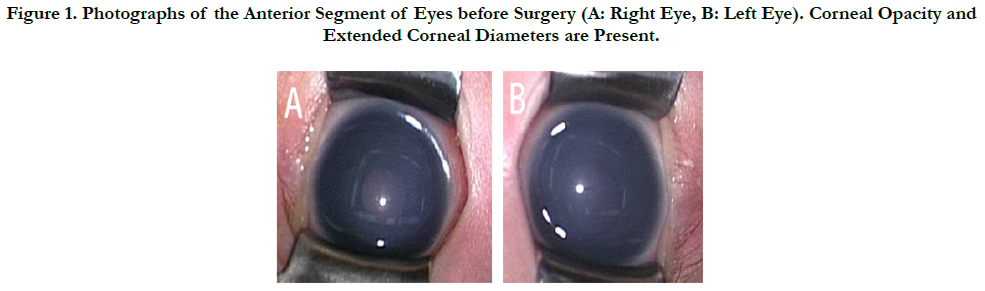
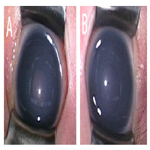
Figure 1. Photographs of the Anterior Segment of Eyes before Surgery (A: Right Eye, B: Left Eye). Corneal Opacity and Extended Corneal Diameters are Present.
At her first visit to our clinic, a slit-lamp examination revealed hazy corneas, round pupils, but not iris atrophy. Her corneal diameter was 11mm (horizontal)×10mm (vertical) OD and 11.5 mm (horizontal)×10mm (vertical) OS. Gonioscopic findings show ednumerous of adherent iris strands on Schwalbe’s line. IOP was 35 mmHg OD and 28 mmHg OS by Tono-Pen AVIA® (Depew, NY), a handheld tonometer, under sedation. Both eyeballs had an axial length of 17 mm. Fundus examination revealed no apparent enlargement of optic disc cupping in either of her eyes.
She underwent trabeculotomy in the temporal inferior area of both eyes under general anaesthesia on Day 22 after birth. In the pre-surgical examination, IOPs were 39 mmHg OD and 37mmHg OS (Tono-pen AVIA®). Because the first surgery for glaucoma had not been effective (IOP levels remained unchanged in both her eyes), she underwent trabeculotomy for the second time in the nasal inferior area of both eyes on Day 36 after birth. After the second surgery, her IOPs stabilized at 11-12 mmHg OD and at 12-14 mmHg OS (Tono-Pen AVIA®) under sedation.
She had a saddle nose and isolation of both eyes. Her father had undergone glaucoma surgery once in both his eyes at the age of 3 months. He had post-operative ventricular septal defect and impaired hearing. Both the patient and her father underwent genetic tests for a definitive diagnosis of DWM, which revealed that both of them carried a mutation of FOXC1.
Ophthalmic examination on her father’s eyes revealed clear corneas and iris atrophy (Figure. 2). His IOP was 15mmHg, the cupto- disc ratio was 0.3, and the visual field test was within normal limits, in both his eyes. His congenital glaucoma was thought to be caused by ARS. The patient was diagnosed with ARS, complicated with DWM based on the result of genetic tests, although her iris was not atrophic at that time. These individuals are therefore suspected of being familial cases of ARS carrying a FOXC1 mutation.
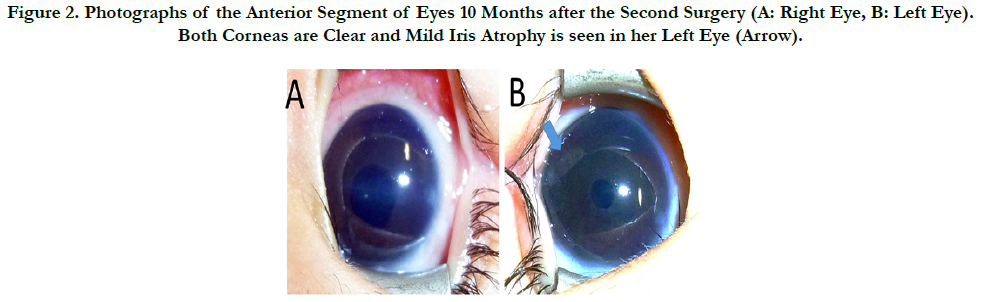
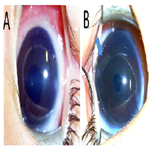
Figure 2. Photographs of the Anterior Segment of Eyes 10 Months after the Second Surgery (A: Right Eye, B: Left Eye). Both Corneas are Clear and Mild Iris Atrophy is seen in her Left Eye (Arrow).
The patient is currently three and half years old; her IOP levels are around the low teens, and both eyes have clear corneas, although mild iris atrophy has been detected in her left eye (Figure. 3). This iris atrophy appeared approximately at the age of 1 year.Fundus examination revealed that the cup-to-disc ratio is 0.5 OD and < 0.4 OS. She has a mild developmental delay.
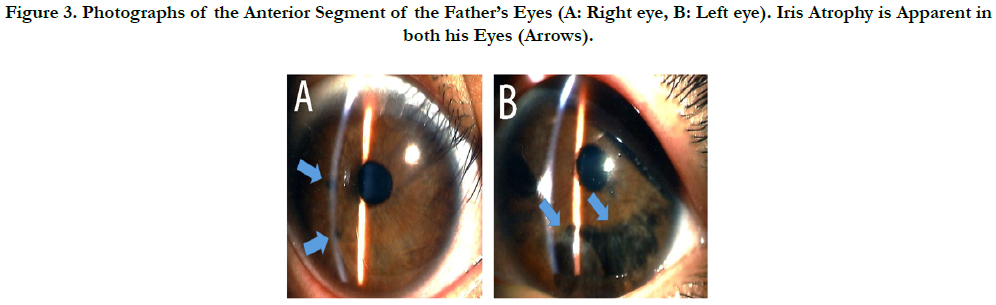
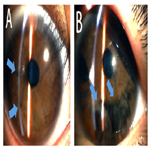
Figure 3. Photographs of the Anterior Segment of the Father’s Eyes (A: Right eye, B: Left eye). Iris Atrophy is Apparent in both his Eyes (Arrows).
Discussion
This is a report of a rare case of a patient with DWM complicated by ARS. Both the patient and her father had a mutation of FOXC1, although her father had ARS without DWM. FOXC1 deletion, duplication, and mutations are reportedly associated with Axenfeld-Rieger anomaly and with DWM [8, 11]. Various combinations of ocular and cerebellar malformations have been found in three fetuses with 6p25 deletions encompassing FOXC1 [11]. Considering these facts, our patient shows that DWM and ARS can coexist.
This patient required trabeculotomy twice to control IOP. On the contrary, her father’s first surgery (trabeculotomy) was sufficiently effective. These different clinical courses might suggest that the disease is more severe in the patient than in her father. Kawase et al. reported 6 Japanese ARS cases in 4 families with FOXC1 mutations [13]. In 3 of the 4 families, a FOXC1 mutation was present in the mothers of the index cases, who had corectopia and iris hypoplasia, but without glaucoma. This report indicated that younger generations may have more severe symptoms than their parents’ generation [13]. This feature is similar to our case.
At her first examination, our patient did not have typical ophthalmological changes of ARS, such as iris atrophy and pupil deformation. Therefore, we did not diagnose her with glaucoma caused by ARS. During follow-up, she underwent genetic tests for definitive diagnosis of DWM, which revealed a mutation in FOXC1 genes. In addition, iris atrophy and pupil deformation appeared in her left eye thereafter. We have therefore changed her diagnosis to that of ARS complicated with DWM. Our case indicates that genetic tests are useful for diagnosing patients with ARS as well as for predicting their prognosis.
Conclusion
Because some ARS cases may not show typical findings, a detailed family history, repeated ophthalmic examinations, and genetic tests are useful for making a definitive diagnosis and predicting prognosis.
Acknowledgement & Declarations
We would like to thank Edit age for editing this manuscript. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophtalmol Soc. 1983;81:736-784. PubMed PMID: 6676983.
- Alward WLM. Axenfeld –Rieger syndrome in the age of molecular genetics. Am J Ophtalmol. 2000 Jul;130(1):107-15. PubMed PMID: 11004268.
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, et al., Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. 1996;Nat Genet. 1996 Dec;14(4):392-399. PubMed PMID: 8944018.
- Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, et al., The forked transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998 Jun;19(2):140-7. PubMed PMID: 9620769.
- Tümer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009 Dec;17(12):1527-39. PubMed PMID: 19513095.
- Millen KJ, Gleeson JG. Cerebellar Development and Disease. Curr Opin Neurobiol. 2008 Feb;18(1):12-9. PubMed PMID: 18513948.
- Grinberg I1, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of thelinked genes ZIC1 and ZIC4 isinvolved in Dandy-Walker malformation. Nat Genet. 2004 Oct;36(10):1053-5. PubMed PMID: 15338008.
- Aldinger KA1, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al., FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009 Sep;41(9):1037-42. PubMed PMID: 19668217.
- Basel-Vanagaite L, Raas-Rotchild A, Kornreich L, Har-Zahav A, Yeshaya J, Latarowski V, et al., Familial hydrocephalus with normal cognition and distinctive radiological features. Am J Med Genet A. 2010 Nov;152A(11):2743-8. PubMed PMID: 20979187.
- Zanni G, Barresi S, Travaglini L, Bernardini L, Rizza T, Digilio MC, et al., FGF17, a gene involved in cerebellar development, is downregulated in a patient with Dandy-Walker malformation carrying a de novo 8p deletion. Neurogenetics. 2011 Aug;12(3):241-5. PubMed PMID: 21484435.
- Delahaye A1, Khung-Savatovsky S, Aboura A, Guimiot F, Drunat S, Alessandri JL, et al., Pre- and postnatal phenotype of 6p25 deletions involving the FOXC1 gene. Am J Med Genet A. 2012 Oct;158A(10):2430-8. PubMed PMID: 22903608.
- Descipio C, Schneider L, Young TL, Wasserman N, Yaeger D, Lu F, et al., Subtelomeric deletions of chromosome 6p: molecular and cytogenetic characterization of three new cases with phenotypic overlap with Ritscher-Schinzel (3C) syndrome. Am J Med Genet A. 2005 Apr 1;134A(1):3-11. PubMed PMID: 15704124.
- Kawase C, Kawase K, Taniguchi T, Sugiyama K, Yamamoto T, Kitazawa Y, et al., Screening for mutations of Axenfeld-Rieger syndrome caused by FOXC1 gene in Japanese patients. J Glaucoma. 2001 Dec;10(6):477-82. PubMed PMID: 11740218